This website is under construction. Please visit after a week for the complete website experience.
Paediatric Surgery, Shaikh Zayed Hospital, Lahore

Choledocal Cyst
Contents
-
Introduction
-
Clinical Presentation
-
Spontaneous Bile Duct Perforations
-
Classification
-
Investigations
-
Management
-
Surgical Procedure
-
Complications and their management
Introduction
The incidence of Choledocal Cyst is 1 in 100,000 to 150,000 and can reach as high as 1 in 35,000. It is more common in Asia with incidence reaching up to 1 in 1000 in Japan. It is more common in females with an incidence reaching 3-4.1:1 (F:M). 60% are diagnosed in the first decade of life, 20% are diagnosed later in childhood/adulthood and 20-25% prenatally.
Majority of Choledocal Cysts are pancreaticobiliary malunion, however PBM can be seen without an associated choledocal cyst in 20-30% of patients and is an independent risk of carcinoma.
Embryology
-
In the 4th week of gestation, hepatic diverticulum forms extending from the ventral aspect of the foregut (endoderm). It divides in to cranial and caudal parts. The cranial part forms the liver and the extra hepatic biliary tree while the caudal part again divides into superior and inferior parts. The superior part forms the gallbladder and the cystic duct and the inferior portion makes the right and left pancreas. In the 6th week, the ventral pancreatic bud and CBD rotate around the duodenum in a 180 degree clockwise fashion and enters at the posteromedial surface of the 2nd part duodenum.
-
In the 7th week, the main pancreatic duct (wirsung's duct) and the CBD junction end in the developing duodenum as closed cavities through elongation to form the ampulla of Vater.
-
In the 8th week, the junction retracts to reside in the submucosa of the duodenal wall, forms a concentric ring in the mesenchyme.
-
By the 12th week, the main pancreatic duct and CBD are obliquely arranged in the duodenum.
-
Arrests at the 12th week of gestation in the ductal plate at the level of the large ducts in the hilum causes Caroli's Disease.
-
Choledococele forms when the CBD and the pancreatic duct open in the duodenum separately or a CBD diverticulum forms in the duodenum.
Possible theory(s):
-
Obstruction of the distal common bile duct including stenosis from biliary inflammation and inspissated bile syndrome.
-
Disordered recanalisation of the biliary system leading to congenital weakness of the wall of the common bile duct.
-
Abnormal innervation of the distal common bile duct resulting in functional obstruction and proximal dilatation.
-
The pancreatic duct joining the common bile duct outside the ampulla of Vater resulting in reflux of pancreatic juice into common bile duct causing dilatation.
Basic Facts:
-
Choledochal cyst type I fusiform is the most common type.
-
Choledochal cyst is more common in females and Asians.
-
More than two-thirds of cases are diagnosed in children less than 10 years of age.
-
The malignancy risk is age related; malignancy mainly affects adults and may develop in the cyst wall or gallbladder.
-
The common hepatic duct and the cystic duct form the base and inferior margin of Calot’s triangle. The superior border was formerly the cystic artery; however, the inferior surface of the right hemiliver is accepted as a better working definition. Endangered structures while dissecting Calot’s triangle include the right hepatic artery, aberrant right hepatic artery and accessory or replaced sectional ducts that join the common hepatic and the cystic duct. The structures within the hepatoduodenal ligament are the common bile duct, hepatic artery and portal vein. The right and left hepatic arteries usually lie posterior to the bile ducts at the liver hilum. The common bile duct contains two main arteries running longitudinally at the medial and lateral aspects of the duct. Intramural resection of the posterior wall of the cyst may help to avoid damage to the portal vein and hepatic arteries if there is dense inflammation or portal hypertension.
Histopathology of the cyst:
Lining of the cyst is not composed of typical biliary mucosa but is rather acellular and lack the typical mucosal lining. Mucin secreting cells are seen in intramural glands. A GI mucosa type composition of immunoreactive gastrin and somatostatin containing cells may also be found in the cyst wall. Due to repeated destruction and regrowth metaplasia can occur: small areas of columnar epithelium and bile ducts can be noted in the cyst wall. Metaplasia is directly proportional to the age and risk of carcinoma.
Histopathology of the liver in long standing Choledocal Cyst:
Mild periportal fibrosis can be seen.
Longstanding cyst causing carcinoma:
Adenocarcinoma is the most common. Small cell carcinoma is the second most common and can occur in the cyst, gallbladder or the pancreas.
Histopathology of Type III Choledocal Cyst
Contains duodenal type mucosa
Most common association
Polycystic Kidney Disease
While all choledochal cysts represent embryologic anomalies, they can be broadly classified as true congenital cysts that are detected prenatally or present in early childhood and acquired cysts that present in older children due to progressive dilatation of the common bile duct secondary to an inflammatory insult. The latter are more likely to demonstrate a long common channel as seen in this intraoperative cholangiogram.
The classic triad of abdominal pain, jaundice, and right upper quadrant mass is seen in only a minority of patients. The diagnosis should be suspected in children with chronic relapsing abdominal pain, recurrent pancreatitis, acute cholangitis without cholelithiasis, and cholelithiasis or biliary sludge without obvious risk factors.
Clinical Presentation
Classical presentation of abdominal pain, jaundice and a palpable RUQ mass occurs in only 20% of cases.
-
Infantile form occurs < 12 months of age and presents with obstructive jaundice, acholic stools and hepatomegaly and signs of hepatic fibrosis. These patients may benefit from early treatment.
-
Adult form occurs >12 months of age and can present with fever, nausea, vomiting and jaundice. It usually occurs after 2 years and can cause mucus plug and biliary sludge.
Perforation of choledocal cyst occurs in about 1 - 12% of patients due to fragile cyst wall, inflammation, increased ductal pressure or increased intraabdominal pressure. Site of rupture is usually the low flow region at the junction of the cystic and the common bile duct. Patient can present with peritonitis, sepsis and abdominal pain.
Long standing choledocal cysts can have chronic inflammation that can cuase mucus or protein plugging and is likely due to inflammation leading to formation of albumin rich exudates or dysplastic epithelium. It can also cause secondary biliary cirhosis in about 40-50% of patients due to obstruction and inflammation causing portal hypertension.
-
Progressive hepatic fibrosis may develop in the infant and is reversible with surgical repair.
-
Recurrent abdominal pain is the dominant presenting feature in older children with or without intermittent jaundice.
-
Portal hypertension may develop in older children because of portal vein obstruction from cyst compression or secondary biliary cirrhosis.
-
Spontaneous rupture may affect any part of the cyst and can be intraperitoneal or retroperitoneal.
Even in the presence of pancreaticobiliary malunion, hyperamylasaemia is not found in infants with choledochal cyst. This is due to pancreatic immaturity and only reaches significant levels at about 1–2 years of age. However, biliary concentrations of pancreatic lipase, elastase and trypsin are significantly elevated in infants with a common channel. Other choices are true.
In infants, choledochal cyst needs to be distinguished from cystic varieties of biliary atresia and other intra-abdominal cysts. Ovarian cysts are usually easily distinguishable by their location in the pelvis. In the rare instance that ovarian cysts reach the size that allows them to extend into the subhepatic space, clinical details allow differentiation from choledochal cysts.
Choledochal cyst presentation has been classified as infantile and adult forms. Infants typically present with obstructive jaundice, with or without acholic stool, due to inflammation and bile stasis. Abdominal mass and vomiting may be noted. It is critical to detect cholestasis in infants and distinguish between surgical and non-surgical conditions. In addition to thorough physical examination, a hepatic panel as well as perinatal infection and metabolic workup should be investigated. Choledochal cyst may be detected by prenatal ultrasound as early as 15 weeks’ gestation. It should be differentiated from other cystic lesions within the area such as duodenal atresia, ovarian cyst, duplication cyst and, most important, cystic variants of biliary atresia. Postnatal scans of progressive enlargement of the cyst or dilatation of intrahepatic ducts are indicative of a choledochal cyst rather than biliary atresia. Affected infants who are otherwise well should be treated with early surgery to exclude biliary atresia and prevent further complication including hepatic fibrosis, cholangitis, rupture and portal hypertension. Hepatobiliary scintigraphy with technetium-99m is widely used in addition to abdominal ultrasound to provide more information in terms of function of the hepatobiliary system. A typical scan shows a filling defect in the liver followed by an increase in radioactivity uptake in the cyst. A 24-hour postinjection scan may be necessary in a jaundiced patient with obstructed biliary drainage. Plain abdominal radiography is useful in distinguishing intestinal obstruction and a routine investigation for most acute abdomen patients. Occasionally, large choledochal cysts with inflammation may depict soft tissue density on plain radiography displacing the surrounding intestines to the left.
Clinical manifestations in the adult form generally do not become evident until after 2 years of age. Infants typically present with obstructive jaundice, vomiting, fever, failure to thrive and abdominal mass. Abdominal pain is the main presenting symptom in older children. It is this age group where the classical triad of jaundice, abdominal pain and right hypochondrial mass may be noted in 5%–10% of patients. Secondary biliary cirrhosis, compression of the portal vein by choledochal cyst or portal vein thrombosis can cause portal hypertension. Patients may present with coagulopathy, splenomegaly, haematemesis or melaena from variceal bleeding. Occasionally, portal hypertension resolves after cyst excision. Rupture can occur in up to 7% of cases and be either intraperitoneal, causing abdominal pain, distension, vomiting, fever and progressive biliary peritonitis, or retroperitoneal, which is less dramatic. At operation it must be differentiated from idiopathic perforation of bile duct with or without associated pseudocyst for defi nitive treatment. Thus, intraoperative cholangiography is recommended. Children with severe, recurrent abdominal pain should have a plasma amylase and/or lipase level measured. A careful detailed anatomical investigation with fasting abdominal ultrasound provides sufficient information about the pancreaticobiliary system and may prompt further investigation. Unexplained pancreatitis in children should be investigated in order to exclude anatomical abnormalities such as a common pancreaticobiliary channel. A dilated long common channel is often associated with choledochal cyst and complicated by acute or chronic pancreatitis from protein plugs and calculi.
Pancreaticobiliary malunion without cystic biliary dilatation has been termed a ‘forme fruste’ type of choledochal cyst. Patients with forme fruste variant of choledochal cyst may be asymptomatic, or occasionally present with obstructive jaundice and recurrent abdominal pain. Associated anomalies include inspissated bile plug, inflammatory stricture and calculi as the causes of obstruction. Although ERCP with sphincteroplasty may seem reasonable, patients with forme fruste should be treated by bile duct disconnection in a similar way to a choledochal cyst because of a high incidence of bile duct mucosal dysplasia from associated long common pancreaticobiliary channel.
Choledochal cyst type III choledochocele is rare, representing only 1.4% of choledochal cysts, and associated with non-specific symptoms varying from asymptomatic to severe pancreatitis. It is usually intraduodenal, but occasionally it is intrapancreatic. The aetiology of choledochocele is thought to be different from other forms of choledochal cyst; it is a simple diverticulum occurring between the ampullary and common bile duct components of the sphincter or a congenital duodenal duplication that arises in the region since its lining is most commonly the duodenal mucosa. The condition should be differentiated from other more common cystic lesions within the area such as duodenal duplication, pancreatic cyst, renal cyst and retroperitoneal teratoma.
Choledochal cyst type IV is divided into two types: type IVA cysts consist of multiple dilatations of the intrahepatic and extrahepatic bile ducts, while type IVB choledochal cysts are multiple dilatations involving only the extrahepatic bile ducts. Caroli’s disease is characterised by segmental cystic dilatation of the intrahepatic bile ducts. The diagnosis depends on demonstrating the continuity of the cystic lesions with intrahepatic biliary tree, which can be done by ultrasonography, CT scan, ERCP, percutaneous transheptic cholangiography (PTC) or magnetic resonance cholangiopancreatography (MRCP). Cholangitis, biliary lithiasis, liver abscess, cirrhosis, portal hypertension and cholangiocarcinoma are its potential complications. The clinical course can be asymptomatic for the first several years or mild symptoms occur infrequently throughout the patient’s life. The majority of symptomatic cases present with recurrent fever, jaundice and/or pain in the right hypochondrium. When it coexists with congenital hepatic fibrosis, it is termed ‘Caroli’s syndrome’; patients may present with portal hypertension and its sequelae such as ascites, splenomegaly and variceal bleeding. Histological intrahepatic bile duct ectasia and proliferation are associated with severe periportal fibrosis. Clinical course may worsen because of repeated episodes of cholangitis with the presence of intrahepatic calculi, abscesses and sepsis. While localised forms confined to one lobe can be treated with hepatic lobectomy, liver transplantation is the only effective modality for diffuse forms. Although a rare disorder, Caroli’s disease should be considered in the differential diagnosis of chronic cholestasis of unknown cause.


The normal length of the pancreaticobiliary common channel (the junction where the bile and pancreatic ducts meet before entering the duodenum) ranges from to , with an average length of approximately . A length greater than 5mm is typically considered an "anomalous pancreaticobiliary junction
Spontaneous Bile Duct Perforations
Spontaneous perforation of the extrahepatic bile ducts should always be considered in infants who develop obstructive jaundice after an initial period of good health or who present with progressive ascites. The majority of cases present within 3 months of birth. Clinical presentation is usually subacute with mild fl uctuating obstructive jaundice and slowly progressive biliary ascites. The typical site of bile duct perforation is at the junction of the cystic and common bile ducts.
The differential diagnosis includes bile duct perforation secondary to choledochal cyst, trauma, necrotising enterocolitis or biliary obstruction from inspissated bile or ampullary stenosis, but the presence of clear bile and absence of bowel content in the ascites is diagnostic of a ruptured bile duct.
Abdominal ultrasound may show a complex loculated collection around the common bile duct that may superfi cially resemble a choledochal cyst. However, at operation the two entities are very distinct. Operative cholangiography is not necessary to diagnose a spontaneous perforation of the bile duct and may in fact enlarge the defect.
For spontaneous perforation, simple external drainage of the bile without further manipulation of the duct is all that is necessary to allow the perforation to heal. The defect seals after a few days, usually without requiring any further surgery.
Classification
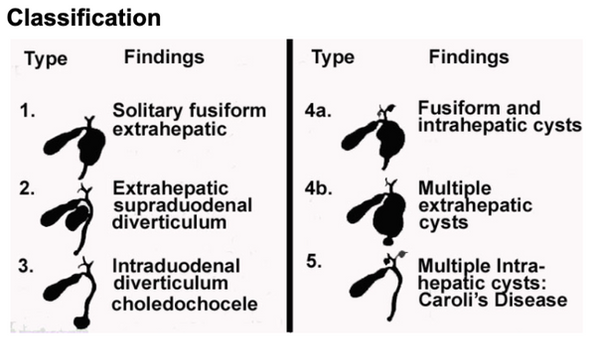
Type 1 is further classified as (a) cystic dilatation of entire CBD (b) Cystic dilatation of a segment of CBD and (c) Fusiform dilatation of the CBD
Investigations
Blood investigations include:
-
Serum markers for obstructive jaundice: conjugated hyperbilirubinemia and increased ALP
Radiographic investigation of choice:
-
MRCP is the gold standard investigation with 90-100% sensitivity (Administration of secretin increases yield by dilating the pancreatic duct and increasing secretion
-
Central dot sign of CT/MRI in Caroli's disease and HIDA scan shows beaded appearance due to intraductal bridging
Prenatal diagnosis can be done in infantile form in the 2nd and 3rd trimester on high resolution USG. Cysts in the porta hepatis (DDs include hepatic cyst, duodenal atresia, mesenteric/omental cyst, intestinal duplication, gallbladder duplication or ovarian cyst)
Investigations Rationale
Ultrasonography is the initial investigation of choice not only to demonstrate the details of the cyst but also the vascular anatomy and hepatic echotexture as well. Stones or sludge in the ducts or extension of the cyst into the liver may be noted. Assessment of biliary anomalies in the liver may also be noted.
ERCP and percutaneous transhepatic cholangiography give excellent visualisation of the duct anatomy. However, both invasive techniques carry a risk of iatrogenic pancreatitis and biliary sepsis, and should be avoided during and immediately after episodes of acute pancreatitis.
Intraoperative cholangiography by injecting into the cyst itself is inefficient and awkward and may fail to fill the intrahepatic ducts. It is best performed through the cystic duct and may require large volumes of contrast to fill out the distal duct including the pancreatic duct and the ducts inside the liver.
CT with intravenous and oral contrast may be used in patients in whom the ultrasound does not provide sufficiently detailed imaging, the cyst is not very dilated, or where an additional problem is suspected. In most patients a detailed ultrasonography supplemented by intraoperative cholangiography provides sufficient information.
Hepatobiliary scintigraphy may be used after surgery to evaluate secretion of bile into the liver but serves almost no purpose in the preoperative workup.
Management
Progressive enlargement of a fetal choledochal cyst with a normal or distended gallbladder or hepatic ducts, or dilatation of the intrahepatic bile ducts demonstrated during pregnancy, favours choledochal pathology. Biliary atresia is not diagnosed prenatally and is rarely associated with large cysts in the hilum of the liver. In biliary atresia, the gallbladder is not typically enlarged and may be undetectable by ultrasound. Hepatic fibrosis may develop rapidly in prenatally detected cases and is known to be reversible with surgical repair. The optimal timing for surgical treatment of prenatally diagnosed choledochal cyst in infants who are otherwise well, is 3 months of age when the baby has established normal feeding patterns, has grown in size and has developed an established pulmonary circulation with resolution of fetal circulatory patterns. The results of surgical treatment at this age are generally excellent.
Spontaneous Rupture
The treatment of spontaneous rupture of the bile duct in a baby includes external drainage with a periductal drain, not an internal drain that may further damage the duct. Ruptured ducts heal spontaneously, after which time the drain can be removed. Long-term care and monitoring with ultrasound is necessary to ensure that the duct heals without stricture formation.
Type I, II and IV
Complete cyst excision and reconstruction by hepaticoenterostomy is the optimum treatment for type I, II and IV choledochal cysts. In type 4 choledocal cysts cyst resection to hepatic ducts leaving in place the dilated intrahepatic ducts because they may decrease in size without distal obstruction. Cystenterostomy may achieve resolution of symptoms in the short term but there is an unacceptable long-term morbidity including malignant degeneration in the cyst mucosa necessitating reoperation and may result in inoperable cancer and death.
Type II choledochal cysts, or common duct diverticula, are quite rare. Some have been misdiagnosed as gallbladder duplications. This anomaly is typically amenable to a laparoscopic diverticulectomy. If a broad communication with the common bile duct is found, a complete common bile duct excision and biliary- enteric bypass, similar to type I cysts, should be performed
In cases of severe cholangitis and severe adhesions
Temporary drainage via T-tube is occasionally required in complicated cases such as patients with ruptured choledochal cyst or intractable cholangitis, which should be easily distinguished from spontaneous rupture of the bile duct. Emergency decompression may allow the cholangitis to resolve and the cyst to be excised at a later time.
Choledochoceles
Large choledochoceles should be removed transduodenally whereas smaller choledochoceles can be treated by sphincteroplasty or endoscopic sphincterotomy with concomitant cholecystectomy. Transduodenal marsupulisation is also an option.
Intrahepatic and Extrahepatic Cysts
Combined cysts in type IV are particularly prone to developing postoperative intrahepatic calculi. Thus a wide Roux-loop anastomosis to the hepatic duct bifurcation is mandatory to facilitate biliary drainage. The hepaticoenterostomy may need to be extended to incorporate hilar duct strictures if present. Debris and calculi should be cleared from dilated intrahepatic ducts as well.
The common hepatic duct is transected just below the level of the confluence of the left and right hepatic ducts providing a single orifice that has not been involved in the cyst itself.
The distal common bile duct is divided just outside the pancreas and oversewn with absorbable suture. It is not followed down through the pancreas, to avoid injury to the pancreatic duct. Operative cholangiography is always advisable since it demonstrates the pancreaticobiliary junction, the anatomy of the intrahepatic ducts and provides a guide to the distal level of bile duct transection necessary. It also shows the position of the individual hepatic ducts in relation to the hepatic end of the cyst where the cyst must be transected without injuring anomalous hepatic ducts. The common hepatic duct is transected at the level of the bifurcation and cholecystectomy should always be performed despite the cystic duct being normal in size.
Choledocal Cyst rupture
Primary external T-tube drainage is helpful in patients with cyst rupture or uncontrolled cholangitis where cyst excision must be performed subsequently.
Complications if cysenterostomy
High rate of anastomotic strictures
Recurrent ascending cholangitis
Bowel obstruction
Portal HTN
Malignancy (epithelial hyperplasia + metaplasia: ERAS gene mutation: later progression of adenocarcinoma of biliary tract: inactivation of DPC gene)
Surgical Procedure for Choledocal Cyst
Roux-en-Y Hepaticojejunostomy
Principles of Surgery
-
Complete excision of the cyst to prevent cholangitis, pancreatitis and malignant transformation
-
Ensure that no abnormal choledocal cyst remain
-
Restoration of biliary enteric continuity using a tension free wide mucosa to mucosa hepaticojejunostomy
-
Ensure free drainage to prevent bile stasis
-
Careful dissection and prevent injury to portal vein.
Surgical Steps
-
Get informed consent and anaesthesia fitness
-
Supine position.
-
Incision: Transverse or Oblique incision in right upper quadrant, open peritoneum
-
Observe: liver, cyst, spleen, biliary channel
-
Get sampling for ascites and sample for cyst fluid for amylase, lipase and culture
-
Create a plane between the peritoneum and the anterior cyst wall. Start dissection inferiorly and continue dissection medially and laterally. Decompress the cyst if needed.
-
Continue the dissection all around till the bile duct tapers towards the duodenum. Encircle and sling it.
-
Mobilize gall bladder, cystic duct and ligate cystic artery.
-
Not oversew the distal bile duct with long acting absorbable suture.
-
Cyst and gallbladder lifted forward to expose the portal vein. Sometimes the right hepatic artery crosses in front of the cyst, take care and dissect it away from the cyst wall.
-
Continue the dissection along the posterior wall taking care of the portal vein. If very adherent and large, then do intramural dissection and seperate the mucosa and the inner wall. Continue dissection submucosally leaving the posterior cyst wall without the mucosa on the vessels (lilly's procedure)
-
Divide the CHD at the level of bifurcation
-
Proximal intrahepatic ducts are cleared to debris and if required they can be washed.
-
Left hepatic duct can be incised 5-10mm for a wider anastomosis
-
Divide jejunum 15-20cm from the ligament of trietz. Oversew the distal end and pass through the mesentery of the transverse colon right to the middle colic vessel
-
End to side anastomosis of the CHD to the jejunum with fine absorbable interrupted sutures (PDS 6/0)
-
The proximal stump is anastomosed oblique end to side 30-4ocm of Roux Loop.
-
Mesenteric defects are closed in both small bowel and transverse colon.
Complications & their management
Immediate:
Hemorrhage
Injury to the duodenum, pancreas and pancreatic duct and liver (to prevent it, do a perop cholangiogram)
Early:
Leakage: A bilirubin or amylase level above that found in the serum suggests anastomotic leak.
Cholangitis: The physiologic prerequisites for cholangitis are bile stasis and bacteria.
Adhesions
Pancreatitis
Ascites due to lymphadema
Late
Adhesion obstruction
Cholangitis
Recurrence
Stricture
Carcinoma
Management of strictures
-
Percutaneous transhepatic balloon dilatation:
-
Endoscopic Retrograde Cholangiography (ERC) with Double-Balloon Endoscopy:
-
Access: Due to altered anatomy (e.g., Roux-en-Y), a short double-balloon endoscope is used to reach the anastomosis.
-
Technique: A guidewire is passed through the stricture, followed by a balloon catheter (e.g., 6–8 mm diameter).
-
Inflation: The balloon is inflated (often up to 8 atm) for 30–60 seconds to open the stricture.
-
Management: Endoscopic nasobiliary drainage (ENBD) may be placed to prevent cholangitis.
-


The Future
Robotic Roux en Y Hepaticojejunostomy
Laparoscopic transumbilical single incision technique
Co-injection of Bile and Indocyanine Green for Detecting Pancreaticobiliary Maljunction of Choledochal Cyst
After dissecting the dilated common bile duct, a percutaneous silicon catheter was inserted into the gallbladder, and bile juice was aspirated. Bile juice and ICG (Diagnogreen; Daiichi Sankyo, Tokyo, Japan) were mixed with the same concentration of ex vivo experiment and co-injected into the gallbladder through the catheter ( Fig. 2A ). The dilated common bile duct and pancreas were detected by NIR fluorescence imaging using a near-infrared/ICG camera (KARL STORZ). This imaging technique was useful for detecting the dissection margin of the distal side of the choledochal cyst inside the pancreatic tissue and preventing injury of the pancreatic tissue ( Fig. 2B ). Injury of the pancreatic duct and pancreas would be recognized as leakage of fluorescence fluid macroscopically if such an injury had occurred.

Radiology Atlas



This baby is not asymptomatic, as he already has mild obstructive jaundiceon laboratory testing. In addition, even asymptomatic babies with prenatallydiagnosed choledochal cysts have been shown to benefit from earlyresection, which is associated with a lower degree of liver fibrosis. A plan is made to resect the cyst at 4 weeks of age. After operation, An ultrasound performed at 3 months shows complete resolution of hepatic duct dilatation. Annual follow-up with physical examination and liver function testing confirms normal growth and development without any clinical or biochemical evidence of obstructive jaundice. At 5 years of follow-up, the child is completely asymptomatic and thriving.
A cystic mass in the upper abdomen of a male fetus may represent acholedochal cyst, cystic biliary atresia, a hepatic cyst, a cystic lymphangioma,an intestinal duplication, or a renal anomaly. Generally, none of theconditions in the differential diagnoses are likely to require fetalintervention or any alteration in delivery mode or timing. The ultrasoundfindings are quite typical of a choledochal cyst and can be described as anexclamation mark or an ice cream cone, with the gallbladder representingthe cone. Cystic biliary atresia is far less likely given the clear visualization ofthe gallbladder
A follow-up ultrasound may be repeated in 2 weeks to exclude rapid growth. If stable, repeat ultrasound may be done every 4 weeks to monitor the growth of the cyst. Postnatal USG is shown below:


TypeIII choledochal cysts, or choledochoceles, have been typically treated bytransduodenal marsupialization. Currently, ERCP is commonly employed totreat these lesions. The cyst is punctured to create a communication with theduodenum and an endoscopic sphincterotomy is performed.