This website is under construction. Please visit after a week for the complete website experience.
Paediatric Surgery, Shaikh Zayed Hospital, Lahore

Blood Disorders
Contents
-
Thalassemia
-
Splenectomy Preparation
-
Surgical Procedure
-
Followup
-
Recent Advances for Splenectomy
Thalassemia
(AR) Decreased synthesis of globin chains of hemoglobin that leads to decreased hemoglobin causing microcytic anemia.
It is an inherited mutation, carriers are protected against plasmodium falciparum malaria.
Three types of hemoglobin:
Fetal: alpha2gamma2
HbA: alpha2beta2
HbA2: alpha2delta2
Divided into alpha and beta thalassemia based
Alpha thalessemia
Gene deletion, normally 4 alpha are present on chromosome 16
-
1 gene deleted = silent carrier
-
2 gene deleted = mild anemia, increased RBC count
-
Cis deletion = deletion of both genes on the same chromosome and it is associated with risk of severe thalassemia.
-
Trans deletion = 1 deletion occurs on each chromosome.
-
-
3 genes deletion: In fetal life, doesn't cause any problems due to presence of fetal HbF, because 1 alpha gene is sufficient to produce HbF but in post natal life, it presents with severe anemia because beta chains produce tetramers HbH and damage RBCs
-
4 gene: lethal in utero and hydrops fetalis occurs, gamma chains produce tetramers called HbBarts that damage RBCs in utero.
Diagnosis is via Hb Electrophoresis
Beta thalessemia
Point mutations in promoter or splicing sites, it is only expressed in postnatal life because there's no beta chain in HbF.
Two genes are present on chromosome 11. Mutations result in diminished or absent beta chains (B0).
Classification:
B+/B0: Minor beta thalasemia: Mildest form of disease and is usually asymptomatic with increase RBC count. Microcytic, hypochromic RBCs with the presence of target cells on blood smear. Hb electrophoresis shows: (1) slightly decreased HbA, (2) increased HbA2 (5%, normally 2.5%) (3) Increased HbF (1 to 2%)
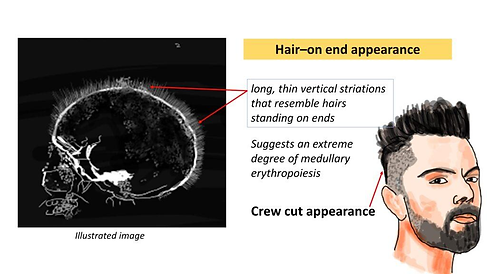
B0/B0: Major beta thalasemia (Cooley's Anemia): Most severe form of the disease and presents as severe anemia few months after birth because high HbF at birth is temporarily protective. After few days, alpha chains form tetramers with damage to RBCs resulting in extravascular hemolysis in the spleen due to removal of structurally defective RBCs. It also causes ineffective erythropoesis resulting in massive erythroid hyperplasia resulting in expansion of hematopoesis in the skull causing frontal bossing. Reactive bone formation causes crew cut appearance on the X ray. It also causes chipmunk facies. Extramedullary hematopoesis occurs with hepatosplenomegaly. There is increased risk of aplastic crisis with parvovirus B19 infection.


Chronic transfusions are necessary and lead to secondary hemochromatosis.
Smear will show: microcytic hypochromic RBCs with target cells and nucleated RBCs.
Electrophoresis will show little or no HbA with increased HbA2 and HbF
Management
As a consequence of extramedullary hematopoesis splenomegally is a frequent finding. Splenic sequestration of RBCs causes enhanced transfusion requirements.
Indications of splenectomy:
-
RBC transfusion requirements exceeding 250ml/kg/year
-
Hypersplenism: Splenomegaly with mono, bi and pancytopenia
-
Massive splenomegaly causing mechanical discomfort
Splenectomy Preperation
Indication: hematologic diseases (sickle- cell disease, hereditary spherocytosis, thalassemia major, and idiopathic thrombocytopenic purpura) and uncontrolled hemorrhage from trauma.
Vaccinations: If possible, vaccinations to Streptococcus pneumoniae, Neisseria meningitidis 5 yearly, and Haemophilus influenzae yearly should be given 2 weeks before the operation.
When removing the spleen electively for disease, a right upper quadrant ultrasound should be performed. If there is gallbladder disease, a cholecystectomy can be done at the same time
Preparation:
Consent and counseling
CBC with peripheral film
LFTs
S/Albumen
PT, aPTT
S/Ferratin levels
Arrange blood
Arrange FFP
Bone marrow
Get echo, do ecg, do ultrasound to rule out cholelithiasis
Anesthesia fitness
Complications
Overwhelming post splenectomy sepsis
Symptomatic splenosis (thrombocytopenia)
Ileus
Acute chest syndrome
Bleeding
Portal vein thrombosis
Post operative management of splenectomy
Advice any type of infection (respiratory) should be promptly treatment
Long acting penicillin (Benzabiotic/Penidura LA) → 0.6/1.2gm strength, one injection once a month life long
Mechanism of anemia in case of thalassemia major
Beta chain of Hb is defective so there is increased sequestration of RBCs resulting in anemia
Indication of splenectomy in case of thalassemia major
Age >5 years, Blood transfusion rate >250ml/kg/year or transfusions 2-3 times a month. Massive splenomegaly
Modes of splenectomy
Open, Laparoscopic
Recent Advances
Ligasure, harmonic scalpel and single port access splenectomy
Partial Splenectomy
Removal of 90-95% of enlarged spleen leaving behind 25% portion of normal spleen size.
Massive Splenomegaly
Length > 17cm or weight > 600g or when the spleen reaches the left iliac fossa
Spleen is only palpable when it is enlarged 2-3times its normal size.
Supramassive splenomegaly
Length craniocaudal >equal to 22cm or weight > 1600g
White pulp
Immune function
Red pulp
Destroys old and defective cells
Surgical Procedure
Goals:
-
Decrease transfusion requirement by 30-50%
-
Decrease iron overload progression
-
Increase Hb, PLT and NEUT count by correcting hypersplenism
-
Prevent complications of massive spleen (pain & rupture)
-
Increase QOL (less fatigue and better growth)
Ideally splenectomy should be done >5 years due to increase chances of OPSI
Principles
-
Patient optimisation: correct anemia, manage iron overload, vaccinate fully
-
Chelation therapy would continue till preoperatively except for defriprone that is to be discontinued 1 week. because it causes agranulocytosis before:
-
Desferoxamine
-
Defresirox
-
Defiriprone
-
-
If patient is not chelated properly, that can cause cardiac arrhythmias.
-
Open if massive
-
Secure vascular control: first ligate artery and then vein to decrease congestion.
-
Look and remove accessory spleen (hilum, pancreas, pelvis, adrenal and testes)
-
Safe dissection.
Preoperative
-
Vaccination, at least 2 weeks before surgery: pneumococcal (5, meningiococcal, hemophilus influenza type b
-
Hb > 10g/dl
-
Continue chelation
-
Labs: CBC for Hb, ferratin and LFTs, USG for splenic size and accessory spleen
Steps
-
Incision: Left upper quadrant transverse incision starting from 11th rib and extending medially or left subcostal incision.
-
Enter the peritoneum
-
Mobilise the spleen
-
Divide the Splenophrenic ligament
-
Splenocolic ligament
-
Ligate short gastric vessels in the gastrosplenic ligament (containing short gastric and left gastroepiploic)
-
Deliver the spleen and pancreatic tail out of the peritoneum and ligate first splenic artery and then splenic vein carefully. If there are varices or splenic vein thrombosis, first ligate splenic artery before hilum and ligamentous dissection.
-
Remove spleen
-
Look greater curvature of stomach and pancreas for injury
-
Look for accessory spleen in pancreatic tail, hilum, kidney, bowel mesentery, gastrosplenic ligament, pelvis and testes.
-
In cases of trauma, if spleen is viable and patient is hemodynamically stable, then do spleenoraphy (with absorbable vicryl mesh wrap all around the spleen with cut for the hilar vessels and stapled on the medial side).
Postoperative
-
Antibiotic prophylaxis: Penicillin G (Penidura LA), 1 monthly injection for 5 years/lifelong 50,000 IU per kg body weight; however not more than 2.4 Million I.U.
-
Early ambulation and breathing exercise
-
Restart iron chelation therapy once wound heals after 1-2 weeks.
-
Continue transfusion program according to schedule
-
Monitor platelet count (reactive thrombocytosis)
-
Autotransfusion syndrome: when spleen shrinks in size, it can cause heart failure due to fluid overload.
-
Monitor for OPSI
Complications:
Immediate
-
Bleeding
-
Infection
-
Thrombocytosis
-
Atelactasis
-
Pneumonia
-
Pancreatitis due to pancreatic tail injury
-
Suppurative abscesses
Early
-
OPSI
-
Venous thrombosis
Late
-
OPSI
-
Persistent hypersplenism
-
Iron overload
Followup
First after 2 weeks to remove stitches and assess wound
then 6 monthly for the first year
then yearly
For regular transfusion schedule
Monitor ferratin - transferrin
Assess development and growth
Booster vaccination
Educate family for OPSI risk factors
Recent Advances for Splenectomy
Laparoscopic Splenectomy
Robotic Splenectomy
Perop navigation and imaging:
NIR flourescence and indocyanine green to localise accessory spleen
Splenic artery embolisation
ERAS
Hereditary Spherocytosis
Most common congenital hemolytic anemia as an autosomal dominant trait.
Abnormal spherical shape due to deficiency of ankyrin leads to abnormal spherical shape of RBC (required for assembly of structural plasma protein spectrin)
Leads to membrance rigidity
Lack of biconcave RBCs leads to trapping and destruction in the splenic pulp
Presents with anemia and jaundice (unconjugated)
infection Can lead to splenic crisis: severe anemia, headache, nausea and abdominal pain.
Long term: growth failure and cholelithiasis
Diagnosis:
Family history
anemic crisis
Peripheral blood smear: spherocytes with increased retic count (15-20%)
negative coomb's test
Osmotic fragility test: normal RBCs rupture in 0.47% N/S but these RBCs rupture at 0.6% N/S

If clinical symptoms are not severe, splenectomy should be deferred till 5 years of age to prevent OPSI
neonates with severe hemolytic anemia, retic count >10%, high bilirubin levels may require urgent splenectomy to prevent kernicterus
USG abdomen should be performed before the procedure to rule out gallstones, if found, cholecystectomy should be performed along with it.
Sickle Cell Anemia
Autosomal Recessive
B chain of hemoglobin, a single aminoacid chain replaces normal glutamic acid (hydrophilic) with valine (hydrophobic)
Protective against plasmodium falciparum
The disease arises when 2 abnormal B genes are present.
HbS (alpha2beta2(s))
in deoxygenated slate HbS polymerises and aggregates into a needle like structure resulting in sickle cell. Increased risk of sickeling occurs with
H - Hypoxemia
A - Acidosis
D - Dehydration

HbF protects against sickeling.
Problems with sickeling:
Two types of sickeling:
Reversible: Sickle and desickle with varying oxygen levels, while passing through microcirculation causing RBC membrane damage and cause intra and extravascular hemolysis, increased risk of bilirubin gallstones. Intravascular hemolysis can lead to decreased haptoglobin and decreased target cells.
Irreversible: lead to vasoocclusion which leads to infarction:
Dactilytis: swollen hands and feets due to vasoocclusive infarcts in bones, common presenting sign in infants
Autosplenectomy: shrunken fibrotic spleen, can cause OPSI, most common cause of death in children, affected children should be vaccinated. Increased risk of salmonella typhi osteomyelitis
Acute Chest Syndrome: Vassocclusion in pulmonary microcirculation leads to chest pain, SOB and lung infiltrates often precipitated by pneumonia. Most common cause of death in adult patients
Pain crisis
Renal papillary necrosis: Results in gross hematuria and proteinuria
Sickle Cell trait: one mutated, one normal B chain, less than 50% HbS. Generally asymptomatic with no anemia. Extreme hypoxia and hypertonicity in medulla can cause sickeling and results in microinfarcts and results in microscopic hematuria and decreased ability to concentrate urine.
Lab:
Sickle cells and target cells seen (Target cells, or codocytes, are abnormal red blood cells that resemble a shooting target (a bullseye) due to an excess of cell membrane relative to their hemoglobin content.)
Howell Jolly Bodies (Howell–Jolly bodies are small, dark-purple, nuclear DNA remnants found inside red blood cells, indicating that the spleen is absent, removed (post-splenectomy), or malfunctioning)
Metabisulfate screening (causes cells to sickle with any amount of HbS) positive in both disease and trait.
Hb Electrophoresis can tell the amount of HbS
Management
Hydroxyurea for sickle cell, painful crisis: induces production of HbF and increased total hemoglobin level and increases MCV. It promotes the release of NO resulting in vasodilation. Dose: oral 20mg/kg/day and increased until the max tolerated dose is reached. Escalate once every 8 weeks. Usually takes 4-8months
Followup. Monthly
L Glutamine: most recent medication
Surgical management:
Avoid hypoxia, hypothermia, dehydration and acidosis, treat infections.
Preop transfusion, Hb > 10g/dl for major procedures, minor procedures can be undertaken safely without transfusion
Previous criteria was to reduce saturation of HbS <30%
Alloimmunisation
Intraop monitoring: Oxygen sat, ABGs, End Tidal, BP, temperature, ECG
Postop: prevent hypothermia, acidosis, dehydration. Assess oxygen saturation and pulmonary status. Fluid and electrolyte balance. Incentive spirometery and adequate hydration and oxygenation.
Anelgesia
Monitor for pulmonary edema and atelectasis.
Acute Abdomen
Painful vasoocculive crisis can present as surgical abdomen. Usually accompanied by an elevated TLC.
Serial examinations
Evaluation by a hematologist.
Acute Chest Syndrome
Rapid transfusion to raise the O2 carrying capacity of blood and dilute HbS
Splenic Sequestration
Defined as the decrease in Hb by 2g/dl below the baseline along with splenic enlargement. Present with acute pain, enlargement of mass in PUQ, increase in nucleated RBC, pallor, decrease in Hb and platelets.
Indication for splenectomy: after one major episode of sequestration, if cholelithiasis then perform a splenectomy
Anesthesia
preop Hb > 10g/dl
adequate blood oxygenation
prevent hypoxia, dehydration, acidosis
Transfusion reaction
Non ABO erythrocyte antibodies develop in 8-50% of patients, increases the risk of reaction. Match E, C and Kell group of antigens.
Adenotonsillectomy
Patients with SCD are at increased risk of developing Obstructive Sleep Apnea due to adenotonsillar hypertrophy (compensatory hyperplasia of lymphoid tissue) and changes in facial bone structures due to chronic hemolysis. Nocturnal hypoxemia can result that can cause painful vasooclusive episodes.
Sickle Cell Anemia
Autosomal Recessive
B chain of hemoglobin, a single aminoacid chain replaces normal glutamic acid (hydrophilic) with valine (hydrophobic)
Protective against plasmodium falciparum
The disease arises when 2 abnormal B genes are present.
HbS (alpha2beta2(s))
in deoxygenated slate HbS polymerises and aggregates into a needle like structure resulting in sickle cell. Increased risk of sickeling occurs with
H - Hypoxemia
A - Acidosis
D - Dehydration
HbF protects against sickeling.
Problems with sickeling:
Two types of sickeling:
Reversible: Sickle and desickle with varying oxygen levels, while passing through microcirculation causing RBC membrane damage and cause intra and extravascular hemolysis, increased risk of bilirubin gallstones. Intravascular hemolysis can lead to decreased haptoglobin and decreased target cells.
Irreversible: lead to vasoocclusion which leads to infarction:
Dactilytis: swollen hands and feets due to vasoocclusive infarcts in bones, common presenting sign in infants
Autosplenectomy: shrunken fibrotic spleen, can cause OPSI, most common cause of death in children, affected children should be vaccinated. Increased risk of salmonella typhi osteomyelitis
Acute Chest Syndrome: Vassocclusion in pulmonary microcirculation leads to chest pain, SOB and lung infiltrates often precipitated by pneumonia. Most common cause of death in adult patients
Pain crisis
Renal papillary necrosis: Results in gross hematuria and proteinuria
Sickle Cell trait: one mutated, one normal B chain, less than 50% HbS. Generally asymptomatic with no anemia. Extreme hypoxia and hypertonicity in medulla can cause sickeling and results in microinfarcts and results in microscopic hematuria and decreased ability to concentrate urine.
Lab:
Sickle cells and target cells seen (Target cells, or codocytes, are abnormal red blood cells that resemble a shooting target (a bullseye) due to an excess of cell membrane relative to their hemoglobin content.)
Howell Jolly Bodies (Howell–Jolly bodies are small, dark-purple, nuclear DNA remnants found inside red blood cells, indicating that the spleen is absent, removed (post-splenectomy), or malfunctioning)
Metabisulfate screening (causes cells to sickle with any amount of HbS) positive in both disease and trait.
Hb Electrophoresis can tell the amount of HbS
Management
Hydroxyurea for sickle cell, painful crisis: induces production of HbF and increased total hemoglobin level and increases MCV. It promotes the release of NO resulting in vasodilation. Dose: oral 20mg/kg/day and increased until the max tolerated dose is reached. Escalate once every 8 weeks. Usually takes 4-8months
Followup. Monthly
L Glutamine: most recent medication
Surgical management:
Avoid hypoxia, hypothermia, dehydration and acidosis, treat infections.
Preop transfusion, Hb > 10g/dl for major procedures, minor procedures can be undertaken safely without transfusion
Previous criteria was to reduce saturation of HbS <30%
Alloimmunisation
Intraop monitoring: Oxygen sat, ABGs, End Tidal, BP, temperature, ECG
Postop: prevent hypothermia, acidosis, dehydration. Assess oxygen saturation and pulmonary status. Fluid and electrolyte balance. Incentive spirometery and adequate hydration and oxygenation.
Anelgesia
Monitor for pulmonary edema and atelectasis.
Acute Abdomen
Painful vasoocculive crisis can present as surgical abdomen. Usually accompanied by an elevated TLC.
Serial examinations
Evaluation by a hematologist.
Acute Chest Syndrome
Rapid transfusion to raise the O2 carrying capacity of blood and dilute HbS
Splenic Sequestration
Defined as the decrease in Hb by 2g/dl below the baseline along with splenic enlargement. Present with acute pain, enlargement of mass in PUQ, increase in nucleated RBC, pallor, decrease in Hb and platelets.
Indication for splenectomy: after one major episode of sequestration, if cholelithiasis then perform a splenectomy
Anesthesia
preop Hb > 10g/dl
adequate blood oxygenation
prevent hypoxia, dehydration, acidosis
Transfusion reaction
Non ABO erythrocyte antibodies develop in 8-50% of patients, increases the risk of reaction. Match E, C and Kell group of antigens.
Adenotonsillectomy
Patients with SCD are at increased risk of developing Obstructive Sleep Apnea due to adenotonsillar hypertrophy (compensatory hyperplasia of lymphoid tissue) and changes in facial bone structures due to chronic hemolysis. Nocturnal hypoxemia can result that can cause painful vasooclusive episodes.
Idiopathic Thrombocytopenic Purpura
Autoimmune production of IgG against platelet antigen IIb/IIIa.
Most common cause of thrombocytopenia
Autoantibodies are produced by plasma cells in the spleen and antibody bound platelets are consumed by splenic macrophages that causes thrombocytopenia
Divided into acute and chronic
Acute: Children, weeks after immunisation or viral infection. Self limited, gets better in weeks to months
Chronic: adults, woman. May be primary or secondary. Can cause thrombocytopenia in offsprings transiently as antibodies cross through the placenta.
Labs.
Decreased platelet counts
Normal PT/aPTT
Coagulation factors are not affected.
Increased megakaryocytes on bone marrow biopsy
Management
Initial treatment is corticosteroids, children respond well. Adults may show early relapse. Perform bone marrow before starting predinisolone and ruling out leukemia as it may be treated partially by prednisolone
IVIG: used to raise platelet counts in bleeding but affect short lived. 0.8-1g/kg IV
AntiD immunoglobulin: can cause brisk hemolysis leading to rare renal failure and death so adequately hydrate before and monitor for hemoglobinurea
Platelet transfusion only indicated in lifethreatening bleeding situation.
Splenectomy eliminates the primary source of antibodies performed in refractory cases.
Idiopathic Thrombocytopenic Purpura
Autoimmune production of IgG against platelet antigen IIb/IIIa.
Most common cause of thrombocytopenia
Autoantibodies are produced by plasma cells in the spleen and antibody bound platelets are consumed by splenic macrophages that causes thrombocytopenia
Divided into acute and chronic
Acute: Children, weeks after immunisation or viral infection. Self limited, gets better in weeks to months
Chronic: adults, woman. May be primary or secondary. Can cause thrombocytopenia in offsprings transiently as antibodies cross through the placenta.
Labs.
Decreased platelet counts
Normal PT/aPTT
Coagulation factors are not affected.
Increased megakaryocytes on bone marrow biopsy
Management
Initial treatment is corticosteroids, children respond well. Adults may show early relapse. Perform bone marrow before starting predinisolone and ruling out leukemia as it may be treated partially by prednisolone
IVIG: used to raise platelet counts in bleeding but affect short lived. 0.8-1g/kg IV
AntiD immunoglobulin: can cause brisk hemolysis leading to rare renal failure and death so adequately hydrate before and monitor for hemoglobinurea
Platelet transfusion only indicated in lifethreatening bleeding situation.
Splenectomy eliminates the primary source of antibodies performed in refractory cases.
VonWillibrand Disease
Autosomal Dominant
Mucosal bleeding
Due to abnormal platelet adhesion and aggragation that arises from subnormal levels of F8 activity and vWF.
Labs
Increased BT
Increased aPTT because vWF is required for the stabilisation of F8
Normal PT
abnormal ristocetin agglutination test
Management
1. Factor 8 and vWF concentrates: treatment of choice to normalise homeostasis
if not available
2, then second option is cryoprecipitate: each unit contains 80-100units of F8 and 80 units of vWF. Dose: provide 10U/kg of F8 equivalent of cryoprecipitate (it is second line as during preperation there is no inactivation of viruses)
3, Desmopressin: Releases F8 and vWF from storage pools in the blood, patients are at risk of hyponatremia and seizures. Monitor perioperative fluid and serum Na.
Hemophilia
Classical Hemophilia: Hemophilia A
X Link Recessive disorders
Decrease levels of Procoagulant factors F8 (A) and F9 (B-christmas factor)
Deep tissue bleeding, hemarthrosis, hematomas and echimosis are common. Recurrent hemarthrosis can cause pseudotumors of bone and hematomas can cause ischemic compartment syndrome
Classification
Severe: Factor levels <1%, give factor prophylaxis in high risk patients to prevent joint problems
Moderate: 1-5%, spontaneous hemorrhage occurs infrequently but relatively minor trauma can cause bleeding in joint and soft tissues
Mild: >5%, rarely have clinical bleeding and only have problem with major trauma, surgical or dental procedures.
Labs
Increased aPTT
Normal PT
Decreased F8 levels
Normal Platelet count and BT
Management
Patient must be screened for presence of an inhibitor to either F8 or F9 during 1-2months before operation.
A low titre inhibitor may be overcome by increased dosage of factors. If high, then it may require the use of (1) Activated prothrombin complex concentrate (F8 inhibitor bypassing activity)
(2) Recombinant activated F7 to bypass the affect of antibody to either F8/F9
On the day of operation:
-
Hemophilia patients recieves a bolus dose of recombinant Factor, usually 50U/kg of F8 bolus in Hemophilia A and a continuous infusion of 4-6U/kg/hour of F8 is started to maintain a factor level of >80% for the next 1-2days (F8 has an half life of 8-12 hours and F9 has a half life of 24 hours). Infusion of 1U/kg of F8 increases plasma level by 2%. Infusion of 1U/kg of F9 increases plasma level by 1%. Factor 9 must be infused in larger amounts to raise the plasma levels.
-
The factor level is checked immediately after the operation and is a final screen for the presence of an inhibitor and attainment of adequate hemostasis.
-
The infusion is reduced on the 2nd or 3rd POD to allow the plasma levels to decrease to 50%
-
Replacement is continued for the next 10-14days. For neurosurgical/orthopaedic procedures continue 4-6weeks especially if physical therapy is planned
-
daily factor levels for the next 3-4 days are necessary and intermittent levels checked after that.
Post Op Management
Perform F8 infusion at home. Lyophilised F8 can be stored in home fridges and have permitted outpatient treatment with self infusions.
Lifelong precautionary measures
Bracelet or disease card should be with the patient
Avoid heavy contact sports
Should not receive NSAIDs
Avoid IM injections
Any minor procedure that requires factor replacement should be combined with major procedures if required to save factor.
Sources
Concentrates
Cryoprecipitate
FFPs
Specific Lypholised F8 concentrates
Recombinant F8/F9 concentrates (Recombinant factors decrease the chances of viral diseases)
How to differentiate between Hemophilia A and coagulation factor inhibitor?
PTT doesnt correct upon mixing normal plasma with patient plasma due to inhibitor. It does correct in Hemophilia A
Vit K deficiency
Disrupts function of multiple coagulation factors. It is activated by epoxide reductase in the liver. Activated Vit K gamma carboxylase activates factor 2,7,9,10, protein C and S.
Deficiency occurs in
1. Newborns (lack of GI colonisation by bacteria that synthesize Vit K - prophylactically given to all newborns at birth)
2. Long term antibiotic therapy (Disrupt Vit K producing bacteria in the Gut)
3. Malabsorption (Deficiency of fat soluble vitamins)
Liver Failure: Decrease production and activation of Vit K
Labs:
Follow PT
Diseminated Intravascular Coagulation
DIC is the pathological activation of the coagulation cascade.
2 problems occur:
1. Widespread microthrombi resulting in ischemia and infarction
2. Consumption of platelets and factors results in bleeding especially from IV sites and mucosal surfaces
Almost always secondary to other disease processes
like
Sepsis: Endotoxin from bacterial wall and cytokines, TNF and IL1. They induce endothelial cells to make tissue factor.
Adenocarcinoma: Releases Mucin that activates coagulation.
AML: Primary granules activates coagulation
Rattlesnake bite: Venom activates coagulation
Obscretic complications: Amniotic fluid (tissue thromboplastin) activates coagulation
Labs:
Increased PT aPTT
Decreased PLT
Decreased Fibrinogen
Elevated D dimers (Fibrin split products) - best screening products
Treatment:
Addressing the underlying cause
Transfusion of blood products and cryoprecipitate