This website is under construction. Please visit after a week for the complete website experience.
Paediatric Surgery, Shaikh Zayed Hospital, Lahore

Biliary Atresia
Contents
-
Introduction
-
Differential Diagnosis
-
Investigations
-
Pathology
-
Procedure
-
Complications
-
Prognosis
-
Follow Up
-
Portal Hypertension
-
The Future
Introduction
It is progressive sclerosing cholangiopathy of unknown etiology
BIliary Atresia occurs in 1 in 10,000 to 1 in 167000 live births with Female:Male ratio to 1.4:1.7.
Types of Biliary Atresia (Japanese Classification)
-
Type I: Level of CBD
-
Type II: Level of CHD (2a: CHD, 2b: CBD, CHD, Cystic)
-
Type III: At the porta hepatis - Most frequent.
Possible theories:
-
Genetic: more in certain populations, associated with other congenital anomalies.
-
Inflammatory
-
Infectious (Virus) - Reo and Rota virus
-
Local ischaemia during fetal hepatobiliary development resulting in abnormal vascular-ductal interactions.
-
Abnormal bile metabolism
-
Pancreaticobiliary maljunction
-
Environmental toxins
60% have pigmented stools sometimes in the postnatal period. Seasonal clustering of biliary atresia in winter months.
Davenport Classification
-
Syndromic Biliary Atresia: Laterality defects and Biliary Atresia Splenic Malformation (BASM) with cardiac defects, inferior vena cava malformations, azygous continuation, preduodenal portal vein.
-
Cystic Biliary Atresia: Cystic change in obliterated biliary tract
-
CMV associated
Composition of bile
97% water
Bile salts
glycorolate
Torocolate
cholesterol
phospholipid
electrolytes
Fatty acids
Proteins
IgA
in the remaining three percents
Bile metabolism
RBC --> Heme --> Bilivirdin (unconjugated bilirubin) --> albumin transport protein to the liver where conjugation occurs by UDP glucoronyl transferase (UGT) converted to conjugated bilirubin --> bile in intestine --> urobilinogen --> converted to urobilin (excreted by urine) and stercobilin (excreted via faeces)
Associated syndromes with BA
MPS CIA
Malrotation
Preduodenal portal vein
Polyspenia
Cardiac malformation
Interrupted IVC
Azygous continuation
Growth failure
is an important consequence of liver disease indicating liver transplantation.
Physiological Jaundice
Jaundice that occurs after 24 hours and before 15 days of life due to breakdown of fetal Hb.
Pathological Jaundice
Jaundice that persists beyond 2 weeks of life and is in the first day of life
What is direct hyperbilirubinemia?
Direct hyperbilirubinemia, since the direct bilirubin is almost six times the upper limit of normal and accounts for 60% of the total bilirubin.
Alagille Syndrome
Alagille Syndrome is a mimicker of BA. The underlying pathology is a paucity of bile ducts. Peripheral pulmonic stenosis is a hallmark of the syndrome. Vertebral anomalies (butterfly vertebrae) and anterior chamber ocular malformations are also seen. The histology of Alagille Syndrome is one of bile duct hypoplasia, rather than bile duct proliferation as seen in BA. However, bile duct proliferation may be seen early in Alagille Syndrome. Therefore, a liver biopsy may not always differentiate the two. A cholangiogram however, would definitively show bile duct patency in Alagille Syndrome. The diagnosis is often suggested early by the presence of associated anomalies. Currently, Alagille Syndrome can be confirmed on genetic testing by identifying the presence of the JAG-1 gene. This gene mutation is present in 97% of cases.
Differential Diagnosis
-
Biliary Atresia (Progressively worsening jaundice, clay coloured stool and dark urine)
-
Bile Puct Paucity Syndrome (Static Jaundice)
-
Inspissated Bile Syndrome (History of delayed passage of meconium and MI)
-
Viral Hepatitis (Fever)
-
Infantile Choledocal Cyst (palpable mass)
The differential diagnosis for jaundice in early infancy is vast and requires a full workup. However, the two main causes of cholestatic jaundice are idiopathic neonatal hepatitis and biliary atresia. Causes of neonatal cholestatic jaundice can be divided into intrahepatic and extrahepatic causes. Extrahepatic causes include biliary atresia, choledochal cyst, bile duct stenosis, strictures and cholelithiasis, spontaneous perforation of the common bile duct, and tumours or masses. Intrahepatic causes are numerous and are generally divided into six possible categories: infectious, metabolic, genetic, toxic, cholangiopathies and others.
Gilbert’s disease is an inherited cause of unconjugated hyperbilirubinaemia. Although it is rare, Gilbert’s disease is the most common genetic cause of unconjugated hyperbilirubinaemia. Haemolytic anaemia is caused by the destruction of premature erythrocytes and is also characterised by unconjugated hyperbilirubinaemia.
The pathology of biliary atresia is characterised by an obstructive biliary pattern that includes fibrosis, inflammation and proliferation. The differential diagnosis for these findings on liver biopsy includes choledochal cyst, bile duct strictures and stones, total parenteral nutrition–associated cholestasis, alpha-1 antitrypsin deficiency, cystic fibrosis, multi drug resistance 3 deficiency, North American Indian childhood cirrhosis (cirrhin deficiency) and Alagille’s syndrome.
Investigations
Blood investigations
-
Liver function tests (Increased conjugated bilirubin, increased ALP, increased GGT [more specific])
-
Clotting cascade abnormalities and abnormal albumin (in case of liver decompensation)
-
Viral markers (Hep B, Hep C, TORCH screening - CMV indicates worse prognosis)
-
Alpha 1 antitrypsin levels
-
Serum Lipoprotein X
Ultrasound Abdomen (4 hours fasting and postprandial to see for the change in the size of the gallbladder lumen, Ultrasound will show contracted gall bladder, triagular cord sign, ghost sign, vascular patency, echotexture of the liver, ascites, status of the spleen)
-
If dilated proximal bile ducts on USG --> Inspissated bile ducts (manage by observation or operative cholangiogram with or without biliary irrigation)
-
Hilar cystic structure --> Cystic biliary atresia or infantile choledocal cyst
-
Triangular cord sign (Hyperechogenic liver hilum between the two portal veins representing fibrotic biliary structures), ghost sign (Atretic Gall bladder, irregular contour, lack of echogenic mucosal lining) --> BA
-
Use of doppler to improve accuracy (echotexture, presence of ascite, patency of hepatic vasculature)
Bedside test to check for biliary atresia
Pass NG tube and aspirate, if yellowish green coloured bile, then biliary atresia is ruled out)
Percutaneous Liver biopsy
Non operative, most accurate way of diagnosing BA. It will show inflammation with ductal proliferation, bile stasis with plugging and giant cell formation.
Intraoperative Cholangiogram
-
Apply a purse string suture on the gallbladder
-
Insert an angiocatheter/laparoscopic cholangiogram catheter/butterfly in the lumen of the gallbladder and tie the purse string suture.
-
Diatrizoic Acid (Hypaque or Gastrograffin) is diluted 1:1 with N/S and injected under real time flouroscopy.
-
If contrast flows freely in the duodenum and into the intrahepatic bileducts then patency has been established --> perform liver bipsy, remove the catheter and close the cholecystostomy.
-
If the liver is fibrotic, nodular with a green colour and the gallbladder is contracted and fibrotic then the diagnosis is confirmed, proceed to Kasai
-
If the gallbladder is normal then put a purse string suture and aspirate, if white bile comes --> diagnosis is confirmed and one can proceed to portal plate dissection without a cholangiogram.
-
If the aspirate is dark and there is ongoing doubt then perform a cholangiogram.
Hepatobiliary Scintography
HIDA: Hydroxyiminodiacitic acid
DISIDA: Diisopropyliminodiacetic acid: More effective in symptomatic cholestasis.
Presence of the isotope in intestine excludes the diagnosis. In liver dysfunction, get a delayed assessment of isotopic secretion after 24 hours.
In jaundiced patients, pretreatment with phenobarbitone 5mg/kg/day for 5 days. It induces hepatic microsomal
This tracer is taken up by the hepatocytes and secreted into the bile. Pictures are taken by a gamma camera.
Kramer Rule
Tells about the probable levels of bilirubin according to the extent of jaundice.
Face (5mg/dl)
Face + Upper trunk (10mg/dl)
Face + Upper trunk + lower trunk + thighs (12mg/dl)
Extending to the arms and legs (15mg/dl)
Going to the soles (>15mg/dl)
Osteopontin, basic fibroblast growth factor and hepatocyte growth factor are associated with biliary atresia progression. A raised level of transforming growth factor-β is associated with improved biliary atresia outcomes.
Screening for cystic fibrosis, hypothyroidism, galactosemia and other metabolic and genetic conditions





Pathology
The characteristic features include expansion of portal tracts, infiltration by inflammatory cells and portal tracts with bile plugs. The expansion of the portal tracts may carry varying degrees of fibrosis that may be predictive of the long-term success of the Kasai procedure. There may also be parenchymal changes such as syncytial giant cell formation, lobular disarray and extramedullary haematopoiesis.
In the pile ducts
Bile duct proliferation, expansion of portal tracts
Bile duct stasis
Varying degrees of fibrosis, infiltration of inflammatory cells
In the liver parenchyma
Syncitial giant cell formation
Lobular disarray
Extramedullary hematopoesis
Limitation: Cannot differentiate between BA and IFLD, alpha 1 antitrypsin deficiency)
Aim of Kasai: After portoenterostomy the ductules responsible for bile drainage, overtime these shoulf form stable bile conduits.
Histological Classification of extrahepatic bile ducts
Type 1 shows absence of bile duct lumen and replacement by a fibroinflammatory cord or tract. Type 2 shows multiple small lumens less than 50 µm surrounding a fibroinflammatory cord or tract. Type 3 shows a distorted, large-sized bile duct lumen surrounded by inflammation and fibrosis.
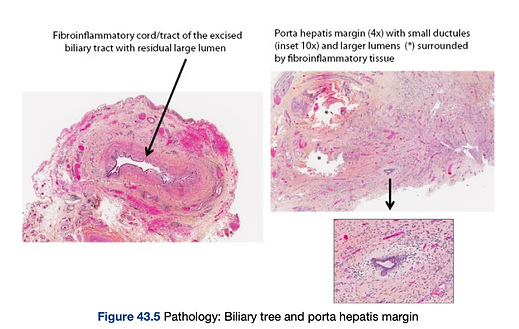
Liver Biopsy
The liver biopsy evaluates the extent of liver injury which is graded on ascale of 1 (portal tract expansion with ductular proliferation) to 4 (cirrhosis).Bile stasis, intrahepatic bile duct loss, giant cell transformation (so-calledgiant cell hepatitis), and portal fibrosis are usually found

Procedure
Preop Preperation:
Optimise Hb, clotting profile,
Vit K
10% Dextrose
PT, aPTT
Blood
Consent and anesthesia fitness
Call for C-arm and flouroscopy
Post Op
IV fluid
Remove NG and start oral after 48-72 hours
Remove abdominal drain at 5th POD or when drainage minimise.
Notice colour of stool, should change immediately or after 10-14 days
NPO for 2-3 days, IV fluids and analgesia.
Start post kasai regime at 3rd to 5th POD
-
IV prednisolone 2mg/kg/day and taber off over 6 weeks (Steroids accelerate clearance of jaundice but donot result in improved survival of the native liver but has chloretic and antiinflammatory properties) - Side effects include: fluid retention and appetite suppression, infections and wound breakdown.
-
Ursodeoxycholic acid 10-15mg/kg/dose x BD for 1 year (it is a chloretic agent that replaces toxic bile acids, increases bile flow and stabilises hepatocyte membrane - it has an antiinflammatory and an antiapoptotic action)
-
Vit ADEK drops 1ml/day for 1 year
-
TP SMX 2.5mg/kg/day
-
Give PPI
Principles:
-
Complete excision of fibrotic extrahepatic biliary remnants upto the porta hepatitis (portal plate)
-
Expose microscopic bile ductules at the porta hepatis to allow bile drainage
-
Create a Roux en Y jejunal limb (classically 40-60cm)
-
Anastomose the jejunal limb to porta hepatis. a wide portoenterostomy reconstruction onto a portal plate, denuded of all tissue.
-
Ensure tension free and water tight anastomosis to prevent leakage and infection.
Incision: RIght subcostal
Perform a cholangiogram. The laparotomy should look carefully for other anomalies (e.g. polysplenia, preduodenal portal vein, absence of the inferior vena cava, and malrotation
-
The liver should be fully mobilized by dividing the falciform ligament, coronary ligaments, and right and left triangular ligaments such that the organ can then be everted outside the abdominal cavity (t is necessary to warn the anesthetist at this stage as the maneuver impairs venous return to the heart by kinking the cava and will need an increase in intravenous volume support.)
-
A stay suture on the gallbladder allows its elevation and this is then separated with bipolar diathermy off its bed. The peritoneum overlying the portal triad (the hilar plate) is then divided and the various vascular (hepatic artery and portal vein) and biliary structures positively identified.
-
The distal common bile duct is ligated and divided and the proximal part elevated from the underlying connective and lymphatic tissue.
-
Following the divided end of the cystic artery should lead to identification of the right hepatic artery, which may be superficial or deep to the common duct and may need slinging
-
Continue dissection proximally along right and left portal veins, extending to: The umbilical fissure on the left. The bifurcation of the right portal vein on the right. Remove all overlying fibrous tissue carefully until a broad raw liver surface is created where microscopic ductules lie. Ensure small oozing points from the raw surface (often indicates good exposure).
-
Identify jejunum about 15–20 cm distal to the ligament of Treitz. Divide jejunum and create: Roux limb: ~30–40 cm (some use 40–60 cm) that will reach the porta without tension. Biliopancreatic limb: remains connected proximally. Jejunojejunostomy: Side-to-side or end-to-side anastomosis between the Roux limb and the distal jejunum, ~30–40 cm distal from division. Ensure good lumen and no twist. Pass the Roux limb: Usually retrocolic (through a window in transverse mesocolon) to the porta hepatis. Check there is no tension and no twist
-
Position the Roux loop snugly against the exposed portal plate. Make a small enterotomy on the anti-mesenteric border of the Roux limb: Length equal to or slightly greater than the width of the exposed portal plate. Posterior row: Interrupted fine absorbable sutures (e.g., 5/0 or 6/0 PDS/Vicryl). Take bites from jejunal mucosa and full thickness of the portal plate capsule (do not go too deep into the liver). Start from one lateral end and go across to the other. Anterior row: Similar interrupted sutures, ensuring: The jejunum is well spread over the whole raw liver surface. No gaps or dead space. Result: a broad mucosa-to-portal-plate anastomosis. Key concept: Extended Kasai = very wide portoenterostomy covering the entire hilar plate, not just a narrow central area.
Original Kasai - Shallow continuous sutures except at 10 and 2 o clock (if needed just place in the connective tissue)
Modified Kasai - Shallow interrupted sutures except at 10 and 2 o clock (if needed just place in the connective tissue)
Extended Kasai - take control of hepatic artery and portal vein on both sides, make a wide anastomosis with deep interrupted sutures through the liver parenchyma even at 10 and 2 o clock




Complications after the procedure
Cholangitis
Occurs due to gram -ve organisms and anaerobes
-
Reflux of intestinal contents
-
Portal venous infections
-
Impaired lymphatic drainage at porta
-
Bacterial translocation
-
Intrahepatic biliary stasis
It causes fever, abdominal pain and jaundice
Treat with antibiotics (TPM + SMX that delays the onset of cholangitis and use of steroid pulses)


Cholangitis testing during hospitalisation
In addition to laboratorytesting, an ultrasound is typically done to assess for any changes, such asintrahepatic bile duct dilatation, fluid around the porta hepatis, or new liverlesions such as cysts or abscesses. These may indicate a problem with apreviously functioning Kasai. The patient should be admitted. Bloodcultures should be sent and broad-spectrum antibiotics should be startedimmediately. The patient should be adequately hydrated. Hemodynamicsand urine output should be monitored. Daily bilirubin and liver functiontests should be checked. A complete blood count should be checkedperiodically during the hospitalization.
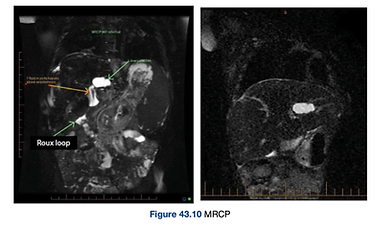
Percutaneous cholangiogram and MRCP post operatively can be used to assess recurrent episodes of cholangitis
It shows an abscess or a bile lake in the left quadrant of the liver. There is good bile drainage through the Roux loop. Definitive treatment of this bile lake is required.
Long bone fractures
Long bone fractures are quite common in patients with BA, occurring in 10%–30% of patients. They are believed to be due to vitamin D deficiency. They can occur with relatively minor trauma and are a common source of morbidity in these patients.

Prognosis
Untreated Biliary atresia is fatal within 2 years
Survival <10% at 3 years
Outcome
pigmented stools immediately or within 10-14 days in 2/3rd cases: half of which will continue to drain bill well, efficient clerance of jaundice and normal development, in other half ongoing fibrosis and scarring causing progressive liver failure = will require liver transplat at 5.4 years
Remaining 15-30% will continue to have acholic stools and liver failure within months
Long term
Abnormalities in menstruation, pubertal disorders Challenges in pregnancy, PTN
Good Prognostic Factors in Kasai
Age < 60 days at the time of Kasai
Gross appearance of gall bladder
Ductules > 110-150micrometer
Experienced surgeon and center
Colic stools within first 2 weeks
Decrease in bilirubin
Type I and II BA
Cystic Biliary Atresia
Worse prognostic factors
BASM
Nodular Liver
CMV associated BA
higher aspartate aminotransferase-to-platelet ratio index (APRi) at time of intervention
Indication of liver transplant:
-
No bile drainage after operation with progressive liver disease
-
Episodic or inefficient bile drainage with slow deterioration of liver function and growth failure
-
Development of one or complications of CLD ie cholangitis, portal HTN that cannot be managed despite functional portoenterostomy
-
>3 months of age,
-
Signs of decompensated liver disease at any age.
Follow up
Follow up at 2 weeks then at 6 weeks till 6 months for
weight
nutritional management
vitamin D levels
LFTs
USG for liver echotexture
Portal Hypertension
34-76% of children even after seemingly excellent results of bile drainage can develop drainage.
Ascites is the most common finding in 60% of children.
Half of the patients will present with esophageal varices so surveillance endoscopy (half yearly to yearly) is recommended and then prophylactic endoscopic sclerotherapy if varices are detected
If varices are bleeding then:
Multiple sessions of sclerotherapy
Octreotide
Beta adrenergic blocking agents
variceal ligation
Variceal bleeding doesnt signify end stage liver disease.
The Future
-
Laparoscopic Kasai
-
Indocyanine green (ICG) near-infrared fluorescence cholangiography has been adapted for Kasai: Helps identify the fibrous cone and residual biliary channels, Guides the level and extent of dissection, Confirms bile flow into the Roux loop intra-operatively.
-
Early diagnosis via stool colour cards and PoopMD app by Johns Hopkins University
-
Modified Kasai + autologous bone-marrow mononuclear cell (BMMNC) infusion by infusing the child’s own bone-marrow–derived mononuclear cells directly into the liver circulation. Paracrine signalling (growth factors, cytokines), Immunomodulation (down-regulating damaging inflammation), Antifibrotic effects (reducing stellate cell activation, collagen deposition), Pro-regenerative effects on remaining hepatocytes